
The entirety of the known universe is teeming with an infinite number of molecules. But what fraction of these molecules have potential drug-like traits that can be used to develop life-saving drug treatments? Millions? Billions? Trillions? The answer: novemdecillion, or 1060. This gargantuan number prolongs the drug development process for fast-spreading diseases like Covid-19 because it is far beyond what existing drug design models can compute. To put it into perspective, the Milky Way has about 100 billion, or 1011, stars.
In a paper that will be presented at the International Conference on Machine Learning (ICML), MIT researchers developed a geometric deep-learning model called EquiBind that is 1,200 times faster than one of the fastest existing computational molecular docking models, QuickVina2-W, in successfully binding drug-like molecules to proteins. EquiBind is based on its predecessor, EquiDock, which specializes in binding two proteins using a technique developed by the late Octavian-Eugen Ganea, a recent MIT Computer Science and Artificial Intelligence Laboratory and Abdul Latif Jameel Clinic for Machine Learning in Health (Jameel Clinic) postdoc, who also co-authored the EquiBind paper.
Before drug development can even take place, drug researchers must find promising drug-like molecules that can bind or “dock” properly onto certain protein targets in a process known as drug discovery. After successfully docking to the protein, the binding drug, also known as the ligand, can stop a protein from functioning. If this happens to an essential protein of a bacterium, it can kill the bacterium, conferring protection to the human body.
However, the process of drug discovery can be costly both financially and computationally, with billions of dollars poured into the process and over a decade of development and testing before final approval from the Food and Drug Administration. What’s more, 90 percent of all drugs fail once they are tested in humans due to having no effects or too many side effects. One of the ways drug companies recoup the costs of these failures is by raising the prices of the drugs that are successful.
The current computational process for finding promising drug candidate molecules goes like this: most state-of-the-art computational models rely upon heavy candidate sampling coupled with methods like scoring, ranking, and fine-tuning to get the best “fit” between the ligand and the protein.
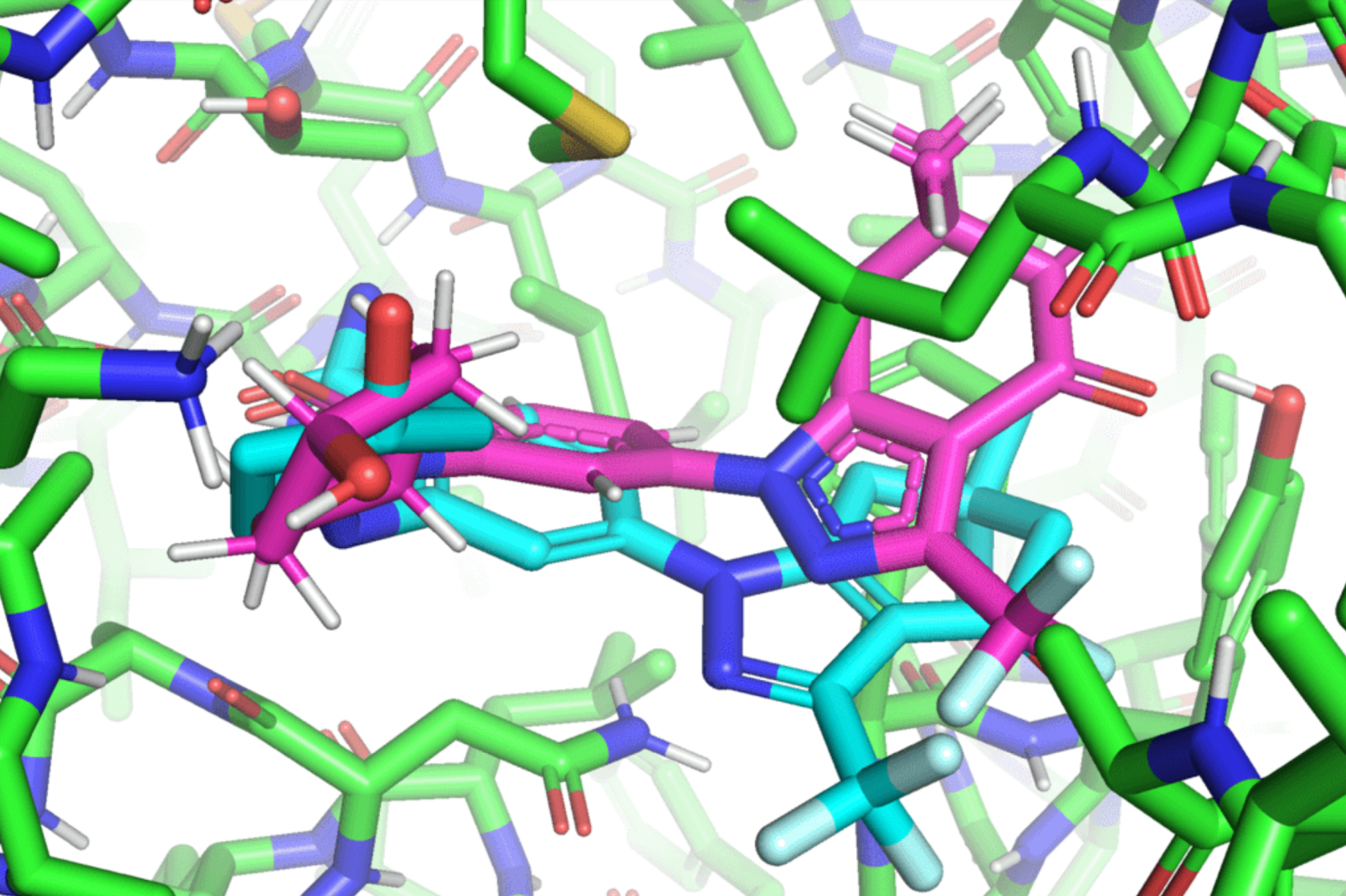
Hannes Stärk, lead author of the paper and a first-year graduate student advised by Regina Barzilay and Tommi Jaakkola in the MIT Department of Electrical Engineering and Computer Science, likens typical ligand-to-protein binding methodologies to “trying to fit a key into a lock with a lot of keyholes.” Typical models time-consumingly score each “fit” before choosing the best one. In contrast, EquiBind directly predicts the precise key location in a single step without prior knowledge of the protein’s target pocket, which is known as “blind docking.”
Unlike most models that require several attempts to find a favorable position for the ligand in the protein, EquiBind already has built-in geometric reasoning that helps the model learn the underlying physics of molecules and successfully generalize to make better predictions when encountering new, unseen data.
The release of these findings quickly attracted the attention of industry professionals, including Pat Walters, the chief data officer for Relay Therapeutics. Walters suggested that the team try their model on an already existing drug and protein used for lung cancer, leukemia, and gastrointestinal tumors. Whereas most of the traditional docking methods failed to successfully bind the ligands that worked on those proteins, EquiBind succeeded.
“EquiBind provides a unique solution to the docking problem that incorporates both pose prediction and binding site identification,” Walters says. “This approach, which leverages information from thousands of publicly available crystal structures, has the potential to impact the field in new ways.”
“We were amazed that while all other methods got it completely wrong or only got one correct, EquiBind was able to put it into the correct pocket, so we were very happy to see the results for this,” Stärk says.
While EquiBind has received a great deal of feedback from industry professionals that has helped the team consider practical uses for the computational model, Stärk hopes to find different perspectives at the upcoming ICML in July.
“The feedback I’m most looking forward to is suggestions on how to further improve the model,” he says. “I want to discuss with those researchers … to tell them what I think can be the next steps and encourage them to go ahead and use the model for their own papers and for their own methods … we’ve had many researchers already reaching out and asking if we think the model could be useful for their problem.”
This work was funded, in part, by the Pharmaceutical Discovery and Synthesis consortium; the Jameel Clinic; the DTRA Discovery of Medical Countermeasures Against New and Emerging threats program; the DARPA Accelerated Molecular Discovery program; the MIT-Takeda Fellowship; and the NSF Expeditions grant Collaborative Research: Understanding the World Through Code.
This work is dedicated to the memory of Octavian-Eugen Ganea, who made crucial contributions to geometric machine learning research and generously mentored many students — a brilliant scholar with a humble soul.
Image and article originally from news.mit.edu. Read the original article here.